SPE-UPLC-MS/MS法測定方便面及其料包中5種罌粟殼生物堿
李坤全,王春雷
(聊城市檢驗檢測中心,山東聊城 252000)
摘 要:選取方便面及其料包為研究對象,建立了固相萃取-超高效液相色譜-質譜聯用法(SPE-UPLC-MS/MS)測定方便面及其料包中5種罌粟殼生物堿的檢測方法,樣品經MCX固相萃取柱凈化,C18柱分離,甲醇和0.1%甲酸水為流動相,梯度洗脫,電噴霧電離正離子模式,多反應監測(MRM)進行定性定量分析,二級質譜進行確證,考察了不同提取液、凈化方式和色譜柱對實驗回收率的影響。結果表明:罌粟堿、那可丁和蒂巴因在0.01~10.0 μg/L,嗎啡和可待因在 0.1 ~ 100.0 μg/L 范圍內線性關系良好,線性系數(r2)> 0.999 8,檢出限為 0.21 ~ 1.24 μg/kg,定量限為 0.66 ~ 4.65 μg/kg,回收率為 72.02% ~ 110.09%,相對標準偏差為(RSD)為 1.05% ~ 4.25%。該方法凈化效果好,檢出限低,回收率高,精密度好,可應用于方便面及其料包中 5 種罌粟殼生物堿的檢測。
關鍵詞:罌粟殼生物堿;固相萃取;超高效液相色譜-質譜聯用;基質效應;方便面
Determination of Five Papaveric Alkaloids in Instant Noodles and its Ingredients Package by SPE-UPLC-MS/MS Method
LI Kunquan, WANG Chunlei
(Liaocheng Inspection and Examination Center, Liaocheng 252000, China)
Abstract: In this research, we selected instant noodles and its ingredients package as research object and established one determination method of five papaveric alkaloids of this object by ultra-high performance liquid chromatography tandem mass spectrometry coupled with solid-phase extraction (SPE-UPLC-MS/MS). The samples were cleaned up by MCX solid-phase extraction column and separated on C18 column using 0.1% formic acid aqueous solution and methanol as the mobile phase with gradient elution, and the target compounds were analysed by electrospray ionization in positive mode with multiple reaction monitoring (MRM) for qualitative and quantitative analysis. The secondary mass spectrometry was used to confirming. This paper also investigated the effects of different extraction solutions, purification methods and chromatographic columns on the experimental recovery. The results suggest that papaverine, noscapine, and thebaine showed a good linearity in the range of 0.01~10.0 μg/L, morphine and codeine also showed a good linearity in the range of 0.1~100.0 μg/L, and the linear coefficient
(2) was >0.999 8. Limits of detections were in the range of 0.21~1.24 μg/kg and limits of quantitations were in the range of 0.66~4.65 μg/kg. The recoveries of alkaloids in instant noodles were from 72.02% to 110.09% with relative standard deviations of 1.05%~4.25%. The method has the advantages of good purification effect, low detection limit, low quantitative limit, high recovery rate and good precision, and can be applied to the determination of 5 kinds of papaverium alkaloids in instant noodles and their packages.
Keywords: papaveric alkaloids; solid-phase extraction; ultra-high performance liquid chromatography tandem mass spectrometry; matrix effects; instant noodles
罌粟殼系成熟罌粟去掉果實之后干燥的果殼,其含有嗎啡(Morphine)、可待因(Codeine)、那可丁(Noscapine)、罌粟堿(Papaverine)和蒂巴因(Thebaine)等生物堿[1],這些成分有斂肺止咳、鎮靜止痛、止瀉等功效[2],但也有一定的成癮性,一些不良商家正是看到了這一點,才鋌而走險,將罌粟殼加入火鍋底料、烹調香料、羊肉湯、麻辣燙、醬鹵肉和飲料等食品中,以此來吸引“回頭客”。如果長期食用這些食品會使人出現虛汗、乏力、面黃肌瘦、精神萎靡等[3]。2009年監管部門規定罌粟殼禁止添加于火鍋食品中,2011年又將禁用的產品類別范圍擴大為“火鍋底料及小吃類食品”,并列出主要成分為“罌粟堿、那可汀、可待因、嗎啡”,從而有效保證人們飲食安全。
目前罌粟殼生物堿的主要檢測方法有光譜法(如分光光度法)、色譜法(如薄層色譜法、氣相色譜法和液相色譜法)、電泳法、免疫分析法、質譜法(如氣相色譜質譜法、液相色譜質譜法)等方法。這其中分光光度法只能對嗎啡定性和半定量測定;薄層色譜法易受干擾;電泳法重現性差,多作為輔助檢測方法;免疫分析法抗體保存時間有限,容易產生交叉反應;氣相色譜法和氣相色譜質譜法不適于難揮發、強極性和易熱解化合物的檢測,而液質聯用法靈敏度、準確度高,精密度好,可進行痕量分析,各組分無需基線分離。
現有文獻報道中研究者已經對醬鹵肉[4]、火鍋底料[5]、羊肉湯及湯料[6]、烹調香料[7]、藥酒[8]和植物油[9]中5種罌粟殼生物堿成分進行了檢測,但是還有一種備受消費者喜愛且容易越吃越上癮的食品——方便面至今沒有檢測報道,故本研究從方便面切入,以固相萃取為凈化手段,結合UPLC-MS/MS建立了方便面及其料包中罌粟堿、嗎啡、那可丁、可待因和蒂巴因5種生物堿的檢測方法,準確度高、檢出限低,二級質譜可有效避免假陽性的出現。
1 材料與方法
1.1 材料
方便面:均來自于政府組織的農村地區流通環節食品安全專項抽檢中的方便面樣品,其中B品牌、J品牌和K品牌各抽出10個,共計30個待測樣品,并按照GB/T 5009.1—2003的要求制備樣品。
1.2 試劑
甲醇(CH3OH,HPLC級)和乙腈(ACN,HPLC級)均購買于OCEANPAK公司;鹽酸(分析純,AR)和氨水(AR)均購買于煙臺遠東精細化工有限公司;正己烷(農殘級):天津市光復精細化工研究所;甲酸(HPLC級):天津市大茂化學試劑廠;乙酸(HPLC級):西隴科學股份有限公司;鹽酸罌粟堿(1 μg/mL)、鹽酸那可丁(1 μg/mL)、蒂巴因(1 μg/mL)、嗎啡(50 μg/mL)、可待因(50 μg/mL)、嗎啡-D3(100 μg/mL)和可待因-D3均購買于MEDJEN LLC公司;MCX固相萃取柱(150 mg,6 mL):美國Waters公司。
1.3 方法
1.3.1 溶液的制備
混合標準工作液:分別精密移取1 μg/mL罌粟堿、那可丁和蒂巴因溶液和50 μg/mL嗎啡和可待因溶液各1.00 mL于10 mL容量瓶中,用甲醇定容至刻度,搖勻,即得含罌粟堿、那可丁、蒂巴因濃度為100 µg/L和嗎啡、可待因濃度為5 mg/L的混合標準品溶液。
混合內標工作液:分別精密移取嗎啡-D3和可待因-D3內標物質(100 μg/mL)各1.00 mL于100 mL容量瓶中,用甲醇定容至刻度,得到嗎啡-D3、可待因-D3的濃度均為1.0 μg/mL的混合內標溶液。
1.3.2 質譜條件
多反應監測模式(MRM),正離子電噴霧離子化(ESI+),離子化電壓為5 500 V,離子源溫度為550 ℃,氣簾氣壓力為35 psi,霧化氣壓力為50 psi,輔助加熱氣壓力為50 psi,各化合物的檢測離子對、碰撞能量(Collision Energy,CE)、去簇電壓(Declustering Potential,DP)、射入電壓(Entrance Potential,EP)和碰撞室射出電壓(Collision Cell Exit Potential,CXP)等質譜參數見表1。
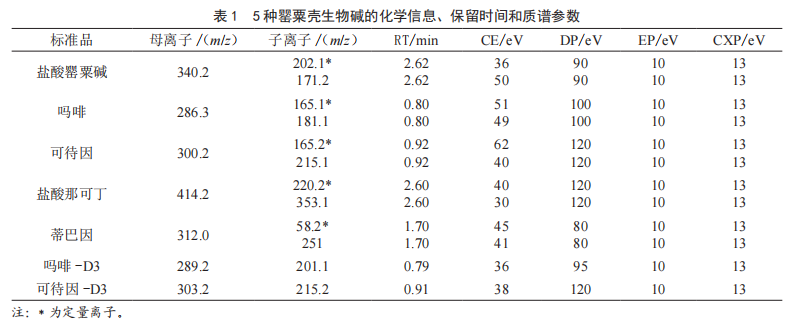
1.3.3 色譜條件
色譜柱:Luna® Omega C18(2.1 mm×100 mm,1.6 µm);流動相;含 0.1% 甲酸水溶液為 A 相,甲醇為 B 相;梯度洗脫程序:0 ~ 1.00 min,70%A;1.00 ~ 3 min,70%A → 10%A;3 ~ 5 min,10%A; 5 ~ 5.1 min,100%A → 70%A;5.1 ~ 6.5 min,70%A;流速:0.4mL/min;進樣量:10 µL。
1.3.4 樣品前處理
準確稱取樣品2 g(精確至0.01 g)于50 mL離心管中,依次加入40 μl內標工作液和20 mL ACN-0.1 mol/L HCl(1∶4,V/V)溶液,渦旋混勻5 min,超聲10 min,低溫(4 ℃)8 000 r/min離心5 min,上清液轉移至50 mL離心管中,加入20 mL乙腈飽和的正己烷,渦旋振蕩2 min,低溫10 000 r/min離心3 min,下層液體待凈化。取MCX固相萃取柱,使用前一次用6 mL水、6 mL ACN活化,準確移取5 mL下清液上柱,保持流速每滴1~2 s,然后依次用6 mL水、6 mL ACN淋洗去除雜質,用6 mL 5%(/)氨水乙腈洗脫,收集洗脫液于15 mL離心管中,40 ℃氮吹至干,加入1.00 mL CH3OH-0.1%甲酸水(3∶7,V/V)定容,過0.22 μm濾膜后上機測試。
2 結果與分析
2.1 前處理方法的優化
5種生物堿均為堿性化合物,酸性環境中易溶于水,因此可用酸性溶液進行提取,提取液用有機溶劑萃取去除脂溶性雜質,再用固相萃取柱進行深度凈化,最后洗脫液旋干復溶過膜上機測定。本實驗分別用 0.1 mol/L HCl 溶液、1% 醋酸溶液、ACN-0.1 mol/L HCl(1 ∶ 1,V/V)溶 液、ACN-0.1 mol/L HCl(1 ∶ 4,V/V)溶液、CH3OH-0.1 mol/L HCl(1 ∶ 1, V/V)溶 液 和 CH3OH-0.1 mol/L HCl(1 ∶ 4,V/V)溶液進行加標回收,結果表明(圖 1),使用 0.1mol/L HCl 溶液和 1% 醋酸溶液進行提取時 5 種化合物的回收率基本上低于 50%,回收效果較差,當使用 ACN-0.1mol/L HCl(1 ∶ 1,V/V)溶 液、CH3OH-0.1 mol/L HCl(1∶1,V/V)溶液和CH3OH-0.1 mol/L HCl(1∶4, V/V)溶液提取時嗎啡的回收效果較差,回收率小于50%,這可能是由于在前處理過程中出現乳化現象,當使用 ACN-0.1 mol/L HCl(1 ∶ 4,V/V)溶液進行提取時回收率均大于 80%,雖有乳化現象的發生,但可以通過低溫高速離心盡量降低損失,故本實驗最終將 ACN-0.1mol/L HCl(1 ∶ 4,V/V)作為提取液。
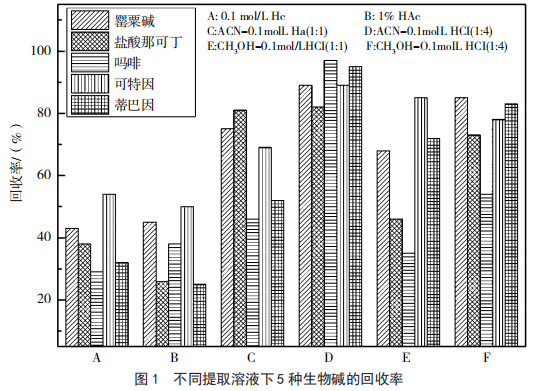
前處理過程中還有一項任務就是盡可能地去除溶劑效應和基質效應,食品樣品種類多,基質復雜,要建立一個適合所有樣品的高效統一的凈化方法實屬不易,因此實驗過程中提取液凈化后氮吹去除乙腈,然后再用初始流動相復溶,以此來減小溶劑效應;同時為更好地降低基質效應,實驗過程中不同品牌的方便面使用不同的基質標準曲線。此外對于凈化方式而言,DB 31/2010—2012[10]和BJS 201802[11]中加無水醋酸鈉、無水硫酸鎂的主要作用是除水和促進生物堿從水相轉移到有機相,提取凈化效率有待研究[12-13];還有學者[14]采用QuEChERS法凈化,此法的確簡便快捷,但是PSA能強力吸附嗎啡,C18可吸附罌粟堿、蒂巴因、嗎啡、可待因導致回收率較低,提取后直接進樣會使基質干擾物增多;固相萃取是較為常見的凈化手段,文獻報道[15]使用C18柱嗎啡無保留,HLB柱嗎啡和那可丁無保留,由于5種生物堿極性較大,故可使用MCX和PCX來凈化,實驗中對比二者的凈化效果,MCX略優于PCX,故最終采用MCX來凈化。
2.2 線性范圍、檢出限和定量限
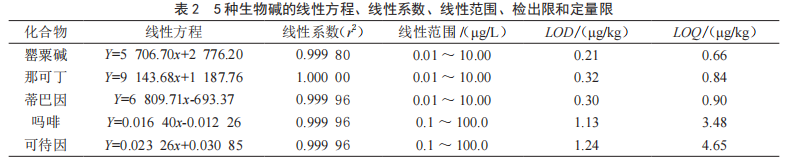
實驗中以基質匹配的方法繪制標準曲線,將空白樣品提取液作為稀釋液,精確配制0.01~10.0 μg/L的罌粟堿、那可丁和蒂巴因的混合標準工作液,0.1~100.0 μg/L的嗎啡和可待因的混合標準工作液(其中含嗎啡-D3和可待因-D3均為10.0 μg/L),各取5 μL上機測試,以標準品濃度為橫坐標,罌粟堿、那可丁和蒂巴因以峰面積為縱坐標,嗎啡和可待因以峰面積和內標物峰面積比值為縱坐標,繪制標準曲線,得到的線性方程、線性系數和線性范圍見表2,結果表明罌粟堿、那可丁和蒂巴因在 0.01 ~ 10.00 μg/L 范圍內,嗎啡和可待因在 0.1 ~ 100.0 μg/L 范圍內線性關系良好,線性系數(r2)> 0.999 8,這表明該法適用于方便面中 5 種罌粟殼類生物堿的檢測。一般以信噪比(S/N) 為 3 的含量定為方法的檢出限(LOD),以信噪比(S/N) 為 10 的含量定為方法的定量限(LOQ),5 種生物堿的檢出限和定量限見表2,檢出限介于0.21~1.24 μg/kg,定量限介于 0.66 ~ 4.65 μg/kg。
2.3 回收率和精密度
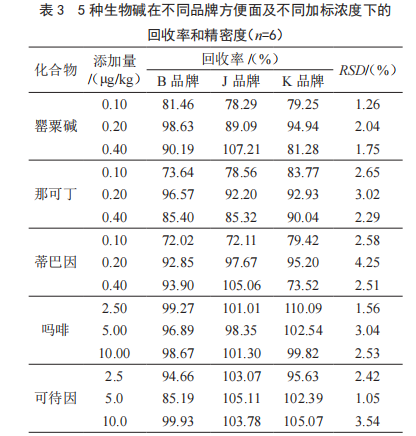
取空白樣品(檢測結果為陰性的樣品),每份稱取2.0 g樣品,分別加入低、中、高3個水平(其中罌粟堿、那可丁和蒂巴因的添加濃度為0.10 μg/kg、0.20 μg/kg和0.40 μg/kg,嗎啡和可待因的添加濃度為2.5 μg/kg、5.0 μg/kg和10.0 μg/kg)的標準溶液適量,每個濃度平行6份,按照1.3.4的方法進行前處理制得待測溶液,上機檢測含量,計算回收率,結果顯示(表3),在3種不同品牌的方便面中5種生物堿的回收率為72.02%~110.09%,相對標準偏差為(RSD)為1.05%~4.25%,說明該方法能夠滿足不同方便面樣品的分析要求。具體來看嗎啡和可待因的回收率(85.19%~110.09%)要明顯優于罌粟堿、那可丁和蒂巴因的回收率(72.02%~107.21%),這可能是因為嗎啡和可待因采用內標法定量,能夠很好地減小基質效應,而其余罌粟堿、那可丁和蒂巴因雖然采用基質匹配標曲外標法定量,但還會或多或少的受到基質干擾,而這一點在3種化合物0.10 μg/kg下的加標回收時尤為明顯,因此以后的實驗中可以采取其他方式(如在線稀釋、開發內標定量法等)近一步降低基質效應。
2.4 二級質譜及其譜庫建立
在實際工作中,由于食品樣品基質復雜,傳統質譜分析的MRM工作模式很容易產生基質效應,導致保留時間和離子比率有偏差,或出現“假峰”,產生假陽性,干擾判斷。為克服此弊端,實驗利用SCIEX復合質譜QTRAP系統獨有的復合掃描模式MRM-IDA-EPI,即三重四極桿和線性離子阱的預掃描方式(Survey Scan)相結合觸發增強離子掃描(EPI),可以在一針進樣的同時獲得MRM色譜峰和增強型二級碎片,其中MRM色譜峰用來定量,EPI不同能量符合二級譜圖形成“指紋”圖譜,實現搜庫和確證,確保檢測結果的準確性。實驗過程中取5種生物堿和兩種內標物溶液單標(濃度為5 μg/L)依次進樣,MRM-IDA-EPI采集模式,將得到的標準譜圖和化合物信息加入數據庫中,建立譜庫并逐一對假陽性樣品進行比對確認。
2.5 實際樣品測定
采用本研究構建方法對政府抽檢的3個品牌的方便面30個樣品進行5種罌粟殼生物堿的定性篩查和定量分析,結果顯示有3個樣品(K品牌2個、B品牌1個)在保留時間1.15 min時嗎啡兩離子對出峰,4個樣品(K品牌2個、J品牌2個)那可丁有一個離子對(414.2/220.2)出峰,4個樣品(J品牌3個、K品牌2個)可待因有一個離子對(300.2/165.2)出峰,考慮到保留時間和離子比率等因素,采集這11個樣品的二級質譜圖,并與標準譜圖進行對比,結果顯示11個樣品3種成分的匹配度(Purity)均小于50%,為陰性樣品。
3 結論
本研究通過使用MCX柱對方便面樣品進行提純凈化,并結合UPLC-MS/MS的MRM和MRM-IDA-EPI采集模式建立了方便面中5種罌粟殼生物堿的分析方法,方法檢出限低,靈敏度高,準確度高,精密度好。采用超高效液相色譜,可在6.5 min內完成一個樣品的分析,大大提高了分析效率,同時建立了5種生物堿的二級質譜圖庫,能有效快速地對陽性樣品進行篩查,確保結果的準確性。本研究不但為罌粟殼生物堿檢測方法國標的制定提供了參考,而且為依法打擊食品中非法添加罌粟殼行為提供有利依據,保障消費者的合法利益。
參考文獻
[1]程月紅,鮑連艷,于艷艷,等.食品中罌粟殼管理現狀及其檢測方法研究概況[J].中國調味品,2018,43(11):196-200.
[2]劉紅,魏柳珍.食品中罌粟殼成分殘留檢測技術及前處理技術研究進展[J].藥物分析雜志,2017,37(10):1747-1753.
[3]管鴻才,陳壽慶.餐飲行業中嗎啡類違禁成分的危害及其鑒定方法研究[J].現代商貿工業,2018,39(22):145-146.
[4]吳義春,孫麗,蘇晶,等.QuEChERS-超高效液相色譜-質譜聯用法測定醬鹵肉中5種罌粟殼生物堿[J].中國食品添加劑,2021,32(4):103-106.
[5]吳瓊,扈明潔,江海,等.分散固相萃取結合通過式固相萃取凈化-超高效液相色譜-串聯質譜法測定火鍋底料中5種罌粟殼生物堿[J].分析科學學報,2021,37(2):205-210.
[6]宋移歡,曹明艷,孫曉紅,等.基于SERS技術快速測定羊肉湯中罌粟殼生物堿[J].食品科技,2020,45(7):378-384.
[7]]睢超霞,陳蕾,徐娜.離子對液相色譜法檢測烹調香料中非法摻雜的罌粟殼[J].中國調味品,2020,45(11):148-150.
[8]陳珉珉,符春花.高效液相色譜-串聯質譜法測定藥酒中5種罌粟殼生物堿[J].
食品安全質量檢測學報,2020,11(16):5776-5781.
[9]楊發震,徐曼曼,蘇少明,等.液液萃取-超高效液相色譜-串聯質譜法測定常見食用植物油中5種鴉片生物堿[J].刑事技術,2021,46(2):146-151.
[10]上海市食品藥品監督管理局.火鍋食品中罌栗堿、嗎啡、那可丁、可待因和蒂巴因的測定 液相色譜-串聯質譜法:DB31/2010—2012[S].上海:上海市食品藥品監督管理局,2012.
[11]國家市場監督管理總局.食品中嗎啡、可待因、罌粟堿、那可丁和蒂巴因的測定:BJS 201802[S].北京:中國標準出版社,2018.
[12]劉敏敏,劉利顏,劉叢叢,等.液質聯用法測定止咳類中成藥中5種罌粟殼生物堿[J].中國衛生檢驗雜,2016(23):3353-3356.
[13]張子臣,李凱,許彤宇,等.液相色譜質譜聯用法測定食品中罌粟殼成分的研究進展[J].食品安全質量檢測學報,2018,9(13):3368-3375.
[14]王力清,酈明浩,李錦清,等.超高效液相色譜-串聯質譜法高通量快速測定調料中罌栗殼生物堿含量[J].食品與發酵工業,2012,38(8):168-172.
[15]顧萬江,周春艷,唐曉琴,等.固相萃取-超高效液相色譜-串聯質譜法同時測定食品中5種生物堿[J].中國衛生檢驗雜志,2014(17):2481-2484.
作者簡介:李坤全(1978—),男,山東聊城人,本科,工程師。研究方向:食品安全與質量。
王春雷(1988—),男,山東聊城人,碩士,工程師。研究方向:食品安全與質量檢測。

相關熱詞搜索:

 探索嬰幼兒輔食市場高質量發展之路,為寶寶成長保駕護航
探索嬰幼兒輔食市場高質量發展之路,為寶寶成長保駕護航
 《食品安全最佳實踐白皮書(2021-2022年)》四大主題發布
《食品安全最佳實踐白皮書(2021-2022年)》四大主題發布
 挪帝克開設京東旗艦店 與京東全球購達成戰略合作
挪帝克開設京東旗艦店 與京東全球購達成戰略合作
 2019《食品安全導刊》雜志訂閱返百元紅包!
2019《食品安全導刊》雜志訂閱返百元紅包!
 9月大事 | 市場監管總局開展2018年全國“質量月”活動
9月大事 | 市場監管總局開展2018年全國“質量月”活動
 盒裝水果省事不衛生 實驗解釋3大疑問
盒裝水果省事不衛生 實驗解釋3大疑問
 使用梅特勒-托利多X光機的五大理由
使用梅特勒-托利多X光機的五大理由
 小個頭 大營養 禾泱泱有機稻鴨原生小泱胚芽米 金
小個頭 大營養 禾泱泱有機稻鴨原生小泱胚芽米 金
 全球食品創新平臺第五期已啟動,攜手共創安全、健康
全球食品創新平臺第五期已啟動,攜手共創安全、健康






參與評論